
生物信息学-蛋白质组学
生物信息学-蛋白质组学
戈亓一、蛋白质组学概述
“蛋白质组学“(Proteomics)和“蛋白质组”(Proteome)是澳大利亚科学家 Marc Wilkins 及其同事在 20 世纪 90 年代初提出的,与 “基因组学”(Genomics)和“基因组”(Genome) 相对应,意为“一个基因组、一种生物或一种细胞、组织所表达的全套蛋白质
- 组学:对生物学和生物体系运作的已汇总全局的研究方式,即从整体的角度来研究一个生物体、细胞或组织。
1.1 基因组学的产生及研究进展
基因组学(Genomics): 研究生物基因组和如何利用基因的一门科学。
- 基因组学分类
- 结构基因组学:全基因组测序
- 功能基因组学:基因功能鉴定
- 基因组学的局限:
- 基因组学推测的大量蛋白功能未知。
- 蛋白功能只能依靠同源基因分析类推和预测
- 一个基因并不代表唯一一种蛋白。
- 一个蛋白在转录、翻译、折叠、修饰中可能有无限变化。
- 蛋白质无法PCR扩增,无法大规模自动化测序。
1.2 蛋白质组学的产生及研究进展
1.蛋白质组学的产生
蛋白质本身的存在形式和活动规律,如翻译后修饰、蛋白质间相 互作用及蛋白质构象等问题,仍依赖对蛋白质的研究来解决。蛋白质组学的研究可望提供精确、详细的有关细胞或组织状况的分子描述。 因为诸如蛋白质合成、降解、加工、修饰的调控过程只有通过蛋白质 的直接分析才能揭示。 因此,蛋白质组学应运而生,它以细胞内全部蛋白质的存在及其 活动方式为研究对象。可以说蛋白质组学研究的开展不仅是生命科学 研究进入后基因组时代的里程碑,也是后基因组时代生命科学研究的核心内容之一。
2.蛋白质组学研究的新进展
主要有五个领域:
- 蛋白质的分离与鉴定
- 蛋白质互作或蛋白质复合体研究
- 蛋白质翻译后修饰分析
- 蛋白质功能鉴定研究
- 蛋白质复合体整体结构分析
二、蛋白质的大规模分离鉴定技术
这种理想的分离方法还应当对不同类型的蛋白质,包括酸性、碱性、 疏水性、亲水性、相对分子质量小、相对分子质量大的蛋白质均能有效分离。
2.1蛋白质二维电泳质谱技术
(一)蛋白质二维电泳技术(2-DE)
利用蛋白质的等电点和分子质量的差异连续进行垂直方向的两次电泳将其分离。
二维电泳和质谱技术联用已成为近年最流行、最可靠的蛋白质分 离与鉴定技术平台。
(二)质谱技术
graph LR; 生物质谱技术 --> id1["基质辅助激光解吸电离 (MALDI),适用于简单的肽混合物"]; 生物质谱技术 --> id2["电喷雾电离ESI 液相分离手段(如液相色谱和毛细管电泳" ]; id1 --> id4["分析器"]; id4 -->fsq1["离子阱(ion-trap,IT)"]; id4 -->fsq2["飞行时间(time of flight,TOF)"]; id4 -->fsq3["四极杆(quadrupole)"]; id4 -->fsq4["傅里叶变换离子回旋共振(Fourier transform ion cyclotron resonance, FTICR)"]; fsq1 --> fsq1you["优点"]; fsq1 --> fsq1que["缺点"]; fsq1you --> 灵敏度高; fsq1you --> 性能稳定; fsq1you --> 具备多级质谱能力; fsq1que --> 质量进度较低; fsq4 --> fsq4you["优点"]; fsq4 --> fsq4que["缺点"]; fsq4you -->高灵敏度; fsq4you -->宽动态范围; fsq4you -->高分辨率; fsq4you -->高质量精度; fsq4que -->操作复杂; fsq4que -->肽段断裂效率低; fsq4que -->价格昂贵;
生物质谱技术在离子化方法上主要有两种软电离技术,即基质辅助激光解吸电离(matrix-assisted laser desorption/ionization,MALDI) 和电喷雾电离(electro-spray ionization,ESI)。MALDI 是在激光脉冲的激发下,使样品从基质晶体中挥发并离子化;ESI 使分析物从溶液相中电离,适合与液相分离手段(如液相色谱和毛细管电泳)联用。 MALDI适于分析简单的肽混合物,而液相色谱与 ESI-MS的联用(LCMS)适合复杂样品的分析。 软电离技术的出现拓展了质谱的应用空间,而质量分析器的改善 也推动了质谱仪技术的发展。生物质谱的质量分析器主要有 4 种:离子阱(ion-trap,IT)、飞行时间(time of flight,TOF)、四极杆(quadrupole) 和傅里叶变换离子回旋共振(Fourier transform ion cyclotron resonance, FTICR)。它们的结构和性能各不相同,每一种都有自己的长处与不 足。它们可以单独使用,也可以互相组合形成功能更强大的仪器。 MALDI 通常与 TOF 质量分析器联用,分析肽段的精确质量,而 ESI 常 与 离 子 阱 或 三 级 四 极 杆 质 谱 联 用 , 通 过 碰 撞 诱 导 解 离 (collisioninduced dissociation,CID)获取肽段的碎片信息。 离子阱质谱灵敏度较高,性能稳定,具备多级质谱能力,因此被 广泛应用于蛋白质组学研究,不足之处是质量精度较低。
与离子阱相似,傅里叶变换离子回旋共振(FTICR)质谱也是一种可以“捕获”离子的仪器,但是其腔体内部为高真空和高磁场环境,具有高灵敏度、 宽动态范围、高分辨率和质量精度(质量准确度可很容易地小于 1mg/L),这使得它可以在一次分析中对数百个完整蛋白质分子进行质量测定和定量。FTICR-MS 的一个重要功能是多元串联质谱,与通 常的只能选一个母离子的串级质谱方式不同,FTICR-MS 可以同时选 择几个母离子进行解离,这无疑可以大大增加蛋白质鉴定工作的通量; 但是它的缺点也很明显,如操作复杂、肽段断裂效率低、价格昂贵等, 这些缺点限制了它在蛋白质组学中的广泛应用。
2.2一维(二维)色谱-质谱技术
(一)一维电泳(色谱)-质谱技术(HPLC-MS)
1.一维 SDS-PAGE 电泳(色谱-质谱技术 一维 SDS-PAGE 电泳结合 MALDI-TOF-MS 技术,或者蛋白质复合物经酶切后用 LC-MS-MS 技术进行分析均被前人的研究证明是有 效的蛋白质分离与鉴定策略。前者的技术流程是先从生物样本中获得蛋白质混合物,然后采用免疫沉淀或免疫亲和的提取方法,获得某种蛋白质的复合体;再应用 SDS-PAGE 技术对蛋白质复合物进行分离、 染色、胶上原位酶切后,用 MALDI-TOF-MS 分析肽质量指纹谱并鉴 定 蛋白质;后者是对蛋白质复合物直接进行蛋白酶切,对获得的肽 混合物进行 LC-MS-MS 分析,通过测定肽片段的序列鉴定蛋白质。
2.一维 IEF 电泳-质谱技术 目前,采用的以 2DE-MS 为基础的蛋白质分析策略,第一维等电聚焦电泳是根据等电点的不同而分离蛋白质,第二维 SDS-PAGE 电 泳是依据相对分子质量的不同再次分离蛋白质。在 SDS-PAGE 电泳 中,可以通过标准分子质量标记物,获得被分离蛋白质的相对分子质量信息。但与质谱技术测定肽段分子质量的方法相比,SDS-PAGE上检测蛋白质相对分子质量的误差大、分辨率差。更重要的是,很难获得翻译后修饰蛋白质相对分子质量变化的准确信息。如何精确测定全细胞蛋白质的相对分子质量,一直是蛋白质组技术方法研究中的难题。 最近,研究人员报道了一种采用 IPG 胶条进行等电聚焦电泳,然后采 用 MALDI-TOF-MS 对胶条进行表面直接分析的方法。
(二)二维色谱-质谱技术(2D-HPLC/MS)
采用液相色谱的方法分离蛋白质和多肽有诸多优点:①速度快, 一般几个小时可完成全部分离过程,而2-DE的蛋白质分离一般需1~ 2d;②由于在溶液状态下,样品处理方便、快速,避免了 2-DE 从胶 上回收样品的繁复操作,分析过程易于自动化和与质谱联接;③对各种蛋白质均适用,包括疏水性、酸性、碱性、分子质量大于 100kDa、 小于 10kDa 的蛋白质等。 从组织、细胞或其他生物样本获得的蛋白质混合物,用蛋白酶(通常是胰蛋白酶)裂解,得到的多肽混合物样品进行二维色谱分离。质 谱分析一般采用电喷雾串联质谱,通过测定肽段序列的分子质量对蛋 白质进行鉴定。二维色谱-串联质谱(2D-HPLC/MS)分析策略的流程 如下图。

二维色谱分离蛋白质有多种组合模式,第一维色谱一般是离子交换色谱,也可用凝胶过滤色谱,第二维通常是反相色谱。由于反相色谱采用有机溶剂作流动相,避免了盐等添加剂对后续质谱分析的影响。 为了提高二维色谱的分离速度和分辨能力,第二维的反相色谱分离系 统一般采用较短的色谱柱和较高的流速。整个分离系统采用一根离子 交换柱与两根反相色谱柱,通过多通道阀进行柱切换的模式操作。当 蛋白质或多肽组分从离子交换色谱柱上洗脱并进入第一根反相色谱 柱时,第二根反相色谱柱上的组分正被洗脱进入检测系统。两根反相 色谱柱的轮流切换,使得被分离物可以富集在柱的顶端。适当调整柱 切换速度与流速,可以达到每分钟一个循环,获得一个色谱图。 减小死体积,采用细内径(0.127mm)的管路并尽可能减少管路的长 度。
荧光差异凝胶电泳技术(difference gel electrophoresis,DIGE)是 对 2-DE 在技术上的改进,结合了多重荧光分析的方法,在同一块胶 上共同分离多个分别由不同荧光标记的样品,并第一次引入了内标的 概念。两种样品中的蛋白质采用不同的荧光标记后混合,进行 2-DE, 用来检测蛋白质在两种样品中表达情况,极大地提高了结果的准确性、 可靠性和可重复性。在 DIGE 技术中,每个蛋白质点都有它自己的内 标,并且软件可全自动根据每个蛋白质点的内标对其表达量进行校准, 保证所检测到的蛋白质丰度变化是真实的。DIGE 技术已经在各种样 品中得到应用,主要用于功能蛋白质组学。
2.3 同位素亲和标签技术 (ICAT)
同位素亲和标签(isotope coded affinity tag,ICAT)技术是近年发展起来的一种用于蛋白质分离与鉴定的新方法。ICAT 方法的关键是 ICAT 试剂的应用。该试剂由 3 部分组成。中间部分被称为连接部 分,分别连接 8 个氢原子或重氢原子,前者称为轻链试剂,后者称为重链试剂。中间部分一头连接一个巯基的特异反应基团,可与蛋白质中半胱氨酸上的巯基连接,从而实现对蛋白质的标记,另一头连接生 物素,用于标记蛋白质或多肽链的亲和纯化。当采用 ICAT 技术进行 蛋白质的定量分离与鉴定时,
2.4 表面增强激光解吸离子化飞行时间质谱技术(SELDI)
表面增强激光解吸离子化飞行时间质谱技术(surface-enhanced laser desorption/ionizationtime of flight-mass spectra,SELDI-TOF-MS) 简称飞行质谱技术。飞行质谱技术的基本原理是,
2.5 蛋白质芯片技术
蛋白质芯片技术(protein microarrays)是一种高通量的蛋白质功能分析技术。
2.6 通过数据库搜索鉴定蛋白质
利用数据库匹配的方式进行肽段鉴定时都遵循一个基本假设,即被研究物种的所有蛋白质序列都是已知并存储于数据库中的。因此, 这种方法只在一些进行了完全测序且基因组注释比较完善的模式生物中应用得比较好,而在非模式生物中的应用则非常有限。 ExPASy(expert protein analysis system) 是由
三、蛋白质的翻译后修饰
蛋白质在翻译合成后,需要经过一些功能基团的共价修饰才能获得相应的活性、结构与功能,这个生物学过程称为蛋白质翻译后修饰 (posttranslational modification,PTM)。
目前已知的蛋白质翻译后修饰类型超过 200 种,常见的类型包括磷酸化、糖基化、泛素化、乙酰化和硫酸盐化等。蛋白质的翻译后修饰通过改变分子大小、疏水性及蛋白质的整体构象等对蛋白质生物学功能的正常行使产生影响。它可以直接影响到蛋白质之间的相互作用,以及蛋白质在不同的亚细胞区域的分布。
3.1 基于序列的蛋白质翻译后修饰预测
利用计算机辅助从蛋白质序列中预测可能的翻译后修饰情况对于蛋白质组数据的生物学注释是非常必要的。发生翻译后修饰的底物蛋白质具有被特定修饰酶所识别的序列基序, 这些序列基序通常只包含 5 个残基,倾向存在于没有固定三维结构的非球形区域(nonglobular regions)内,并且,区域内包含的都是一些有极性的、小的或者骨架柔韧度较大的残基,这样,蛋白质翻译后修饰位点才易于与修饰酶互相接近。但是,如果预测只考虑修饰识别序列基序的话,由于其长度过于短小,就容易在序列模式匹配中产生过多的假阳性。 为了减少假阳性的出现,在预测中除了修饰位点的序列基序特征外,我们还要考虑修饰位点周边环境序列的特点,并尽可能地利用一些统计学方法来增加预测的准确性,如支持向量机(SVM)。
支持向量机(SVM)是一种与线性或二次判别分析相类似的数据分类方法。这种方法将所有的数据映射到一个三维甚至多维空间上,然后利用线性或非线性的函数说形成的最佳超平面将真是的信号与噪声进行区别。简而言之,支持向量机就是将这个修饰位点的预测转化成了以一个分类问题(是或不是修饰位点)。
3.2 在蛋白质组学分析中鉴定翻译后修饰
翻译后修饰也可以通过实验得到的质谱指纹数据进行鉴定。在 ExPASy 蛋白质组学服务器工具页面上列出了很多根据质谱分子质量数据来确定翻译后修饰的程序,可以用来搜索序列中已知的翻译后修饰位点,并且在数据库片段匹配时根据修饰的类型将额外的质量进行整合。
FindMod 可以利用试验确定的肽段指纹信息来比较实际测得的肽段质量与其理论值是否存在差异。可以预测28种不同类型的修饰,包括甲基化、磷酸化、酯化和硫化。
GlyMod是另一种特异性针对糖基化进行预测的程序,它同样是利用实验确定的肽段指纹信息与理论获得的肽段在质量上的差异进行预测。
四、蛋白质分选
除线粒体和植物叶绿体中能合成少量蛋白质外,绝大多数的蛋白 质均在细胞质基质中的核糖体上开始合成,然后运至细胞的特定部位, 这一过程称蛋白质分选(protein sorting),也被称为蛋白质定向转运 (protein targeting)。
蛋白质亚细胞定位的计算预测方法
- 要利用蛋白质的氨基酸序列中的信号特征(signal),同时还需要诸如表达谱数据、系统发育图谱、数据库记录描述内容中的上下文及GO注释词条等额外信息。
- 只利用序列进行对库的同源性搜索或者查找与之共同出现(co-occurrence)的蛋白质结构域。
- SignaIP 是一个基于网络的亚细胞定位信号预测程序。它也是最早使用神经网络进行蛋白质分选信号预测的程序之一。,从第二版 SingalP 开始,预测中又加入了另一种机器学习方法——隐马尔可夫模型来增加预测的准确性。其中, 神经网络算法包含了两种不同的分数,一种是为识别出的信号肽打分, 另一种是为蛋白酶的断裂位点进行打分;而隐马尔可夫模型则用来区分信号肽及将蛋白质插入膜中的 N 端跨膜锚定片段。
- TargetP 是进行真核生物蛋白质亚细胞定位预测的程序。它使用了神经网络方法,主要对蛋白质序列的 N 端是否存在下列几种前导肽进行预测:叶绿体转运肽 、线粒体靶肽(mitochondrial 及 分泌途径信号肽(secretory 。
- PSORT 是一个 1992 年就开发出来的亚细胞定位预测程序,它最先采用了决策树 (decision tree)的方法进行预测,即计算一组来源于定位信号序列的参数并将之与从文献中总结的一系列定位规则进行比较。这些规则大部分关注的是现有的各种能够将蛋白质定位于特定细胞区域的序列基序,还有一部分规则主要记录了特定信号序列基序的氨基酸组成内容。
五、蛋白质相互作用
细胞进行生命活动过程的实质就是蛋白质在一定时空下的相互作用,因此,描绘出蛋白质间的相互作用是蛋白质组学又一个重要的研究内容。蛋白质之间的相互作用不仅包括那些能够形成稳定复合物的强相互作用,也包括那些可能只在瞬时发生的弱相互作用,其中,参与形成复合物的蛋白质要比那些只出现瞬时相互作用的蛋白质表现出更为紧密的表达共调控性质。蛋白质-蛋白质相互作用的研究在探寻细胞信号转导途径、复杂蛋白质结构的建模及理 解蛋白质在各种生物化学过程中的作用方面都是非常必要的。
5.1 利用实验鉴定蛋白质互作
通过实验鉴定蛋白质的相互作用主要包括遗传学方法、生物化学方法和生物物理方法,其结果可以在原子、互作双组分、互作多组分 (复杂互作)及细胞水平等 4 个方面得到表现。
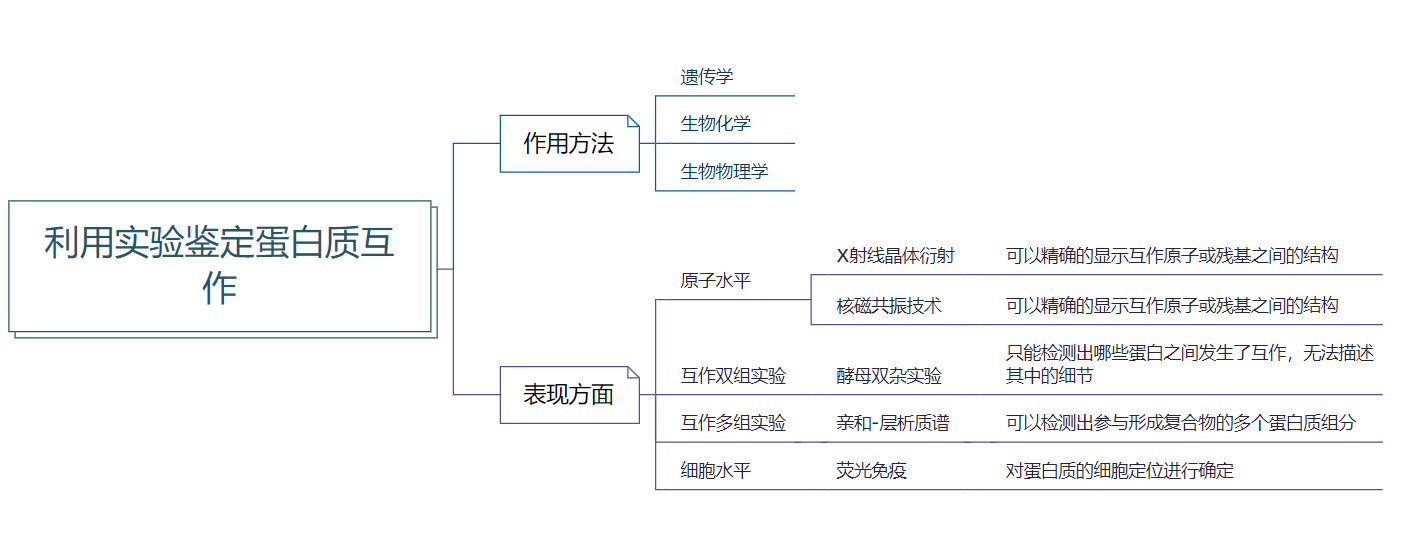

5.2 蛋白质互作的预测
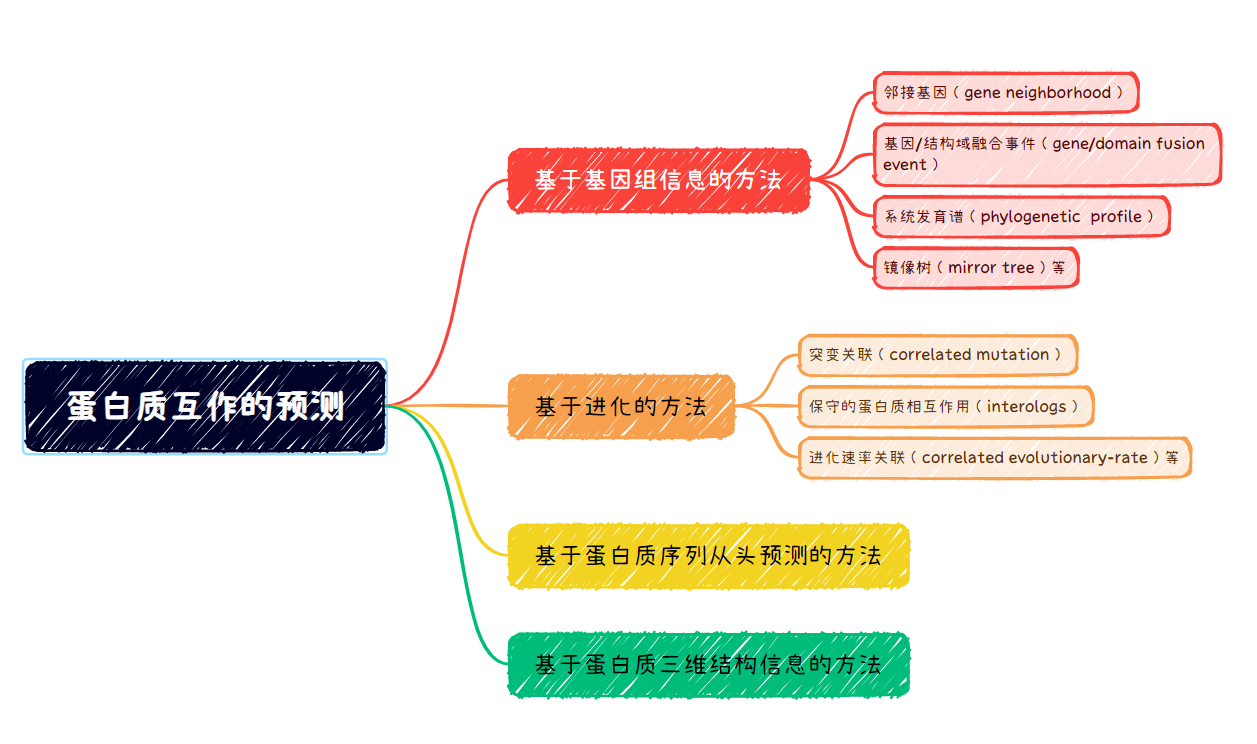
(一)根据邻接基因进行互作蛋白的预测
一般说来,基因次序在不同的原核生物基因组中的保守性很差。 如果确实在不同基因组中发现了具有同样邻接关系的基因,则它们很可能属于同一个操纵子(operon)。这是因为在原核生物基因组中,功能相关的基因承受着相同的选择压力,倾向于在基因组中连在一起,构成一个操纵子。操纵子中编码的蛋白质通常是功能相关或具有物理互作关系的。这种基因之间的邻接关系在物种演化过程中具有一定的保守性,可以作为基因产物之间功能关系的指示标识。这一预测规则支持了大多数原核生物基因组的互作蛋白的预测。但对于真核基因组来说,利用基因次序进行互作蛋白的预测不如基因表达中的共调控蛋白有说服力。
(二)根据基因/结构域融合事件进行互作蛋白的预测
基于基因融合事件的互作蛋白预测方法的理论基础是,对于一个特定物种中包含了融合结构域 AB 的蛋白质家族来说,经常会发现它与其他物种中相互分离的两个基因 A′和 B′相对应。这类蛋白质就被 称为“复合”蛋白(即融合蛋白),而它们包含的每一个结构域都被称为“成分”蛋白。这一预测主要是在具有完整基因组序列的物种中进行基因融合事件的鉴定,因为这样才能发现不同物种中相对应的“同源” 蛋白。如果一个“复合”蛋白特定地与 另一个物种中的两个“成分”蛋白相似,即使这两个“成分”蛋白的编码基因并不一定相邻,但这两个 “成分”蛋白很可能发生相互作用以执行与 AB 相同的功能。

反过来说,如果两个祖先基因 A 和 B 编码了相互作用的蛋白质,那 么在进化的过程中为了加强这种互作的有效性,在其他基因组中这两 个基因很有可能融合在一起。 这一方法更进一步的解释是当两个成分蛋白作为结构域在一个 蛋白质中融合时,它们必须位置非常地接近,以执行同样的功能。如果这两个结构域存在于两个不同的蛋白质中时,为了维持相同的功能, 它们相互靠近及相互作用的特性也必须维持下来。因此,研究基因/蛋 白质的融合事件,就可以对蛋白质-蛋白质的相互作用进行预测。这种利用基因融合事件来揭示其成分蛋白之间的功能相互关系的预测方法被称为“Rosetta stone”方法。这种预测原则被证实是相当可信的,它已经被成功地应用到从原核到真核生物中大量蛋白质互作的预测中。 研究表明,基因融合事件在代谢蛋白(metabolic protein)中尤为普遍,利用此方法进行预测的局限性在于只能够预测在进化过程中发生融合的蛋白质之间的功能关联,不能判断发生融合的蛋白质是否真正发生了物理上的直接接触
(三)根据系统发育谱进行互作蛋白的预测
根据系统发育谱的预测方法就是查找不同基因组中直系同源物共同出现及共同缺失(co-presence and co-absence)的情况
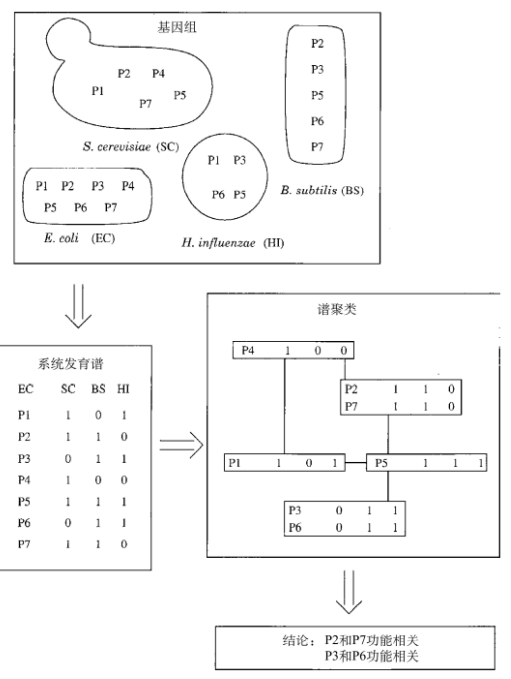
镜像树(mirror tree)是一种基于系统发育分析的更加定量化的互作蛋白预测方法。根据观察,互作蛋白的进化距离相似性水平要显著 高于那些没有互作关系的蛋白质的进化距离,这表明互作蛋白受其功能约束,因而其进化过程应该保持一致。

(四)根据序列的同源性进行互作蛋白的预测
已知的蛋白质互作图谱能够为新测序完成的基因组中蛋白质互作的预测提供必要的信息。如果某个基因组中的一对蛋白质是互作蛋白,那么它们在另一个基因组中的同源蛋白也可能发生类似的互作。 这种同源蛋白对被称为互作物(interologs)。这种预测方法依赖于确定正确的直系同源蛋白并且要利用现有的互作蛋白数据库。
(五)利用多种方法进行互作蛋白的预测
STRING(search tool for the retrieval of interacting gene/proteins, 互作基因/蛋白搜索工具)是一种综合利用了邻接基因、基因融合及系统发育谱等证据来进行蛋白质互作预测的数据库及网络服务器。最新版本的 STRING 除了计算预测外,还整合了大量 实验获得的共表达数据及从文献中挖掘到的蛋白质互作信息。经过分析,STRING 可以给出包括直接及间接的蛋白质-蛋白质相互作用的所有证据。
- 直接互作是指蛋白质之间发生物理互作的情况,而间接相互是指那些功能相关的在同一(代谢)途径中拥有相同底物的酶类或者指在同一遗传途径中相互具有调控关系的蛋白质。
- STRING8 包含了来源于 630 个基因组的 250 万个蛋白质的相关信息。
- 使用 STRING 进行计算预测分析时,查询序列首先会根据 COG (cluster of orthologous group)信息进行直系同源物的分组,然后序列会在数据库中搜索已知的保守基因连锁模式(查找邻接基因)、基因融合情况及系统发育谱。
- STRING 利用一个权重记分系统来评估各 个基因组中上述 3 种类型蛋白质相关的显著性,以及从实验中获得的 互作数据和文献挖掘的信息。为了降低假阳性并增加预测的可信度, 3 种计算类型的相关蛋白质还会在一个内部参考数据集中做再次的检查。
- STRING 的最终结果是返回一个互作蛋白(功能相关蛋白)列表, 针对每一个功能相关蛋白都会提供所得到的计算预测、实验获得及文献挖掘方面的证据,以及一个简单的蛋白质互作(功能相关性)分数。 根据其中参与的多个功能相关伴侣情况,STRING 还能够以交互的形 式提供图形化显示的蛋白质结合互作网络及特定的详细信息







