
生物信息学-序列比对原理
生物信息学-序列比对原理
戈亓一、认识序列
什么是序列
序列(squence) 可以理解为 字符串(string)
- Si 表示序列的第i个元素
- S‘ 代表S的子序列
- 蛋白质序列:由20个不同的字母(氨基酸)组合而成。
- 核酸序列:由4个不同的字母(碱基)排列组合而成
序列常用格式:
FASTA格式
- 第一行:大于号 + 名称 或 其他注释
- 第二行:每行60个字母或80个(根据需要不一定)
二、序列的相似性
序列一致度和相似度
前提:两个序列长度相等
- 一致度:两条序列对应位置上相同的残基(一个字母,氨基酸或碱基)的数目占总长度的百分数。
- 相似度:两条序列对应位置上相似的残基与相同的残基的数目和占总长度的百分比
什么是[残基]:氨基和羧基脱水缩合后剩余的部分
那么 哪个残基与哪个残基算作相似呢?
可以用替换计分矩阵来计算。
三、替换记分矩阵
是反应残基之间互相替换率的矩阵,它描述了残基两两相似的量化关系,分为DNA替换记分矩阵和蛋白质替换记分矩阵
3.1常见DNA序列的替换记分矩阵
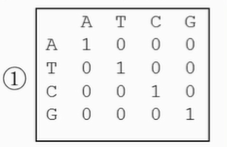
等价矩阵
相同和核苷酸之间的匹配得分为1分,不同核苷酸间的替换得分为0。
实际的序列比较中较少使用
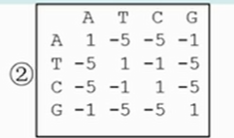
转换-颠换矩阵:
嘌呤(腺嘌呤A,鸟嘌呤G)和嘧啶(胞嘧啶C、胸腺嘧啶T)之间的转换成为颠换

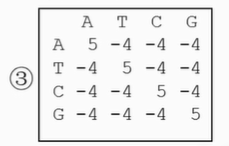
BLAST矩阵

3.2蛋白质序列的替换矩阵
等价矩阵
PAM矩阵
基础的PAM-1矩阵反应的是进化产生的每一百个氨基酸平均发生一个突变的量值,PAM-1自乘n次,可以得到PAM-n,即发生了更多次突变。
BLOSUM矩阵


PAM-X x表示大致的差异度,并非准确的差异度,BLOSUM-X 中x表示序列的相似度,并且等于相似度

如何选择PAM矩阵还是BLOSUM矩阵?
对于关系较远的序列之间的比较,BLOSUM更具优势(PAM是推算而来的,准确度受到一定的限制),对于序列关系较近的序列之间的比较,二者差别不大。
4.遗传密码矩阵
5.疏水矩阵

四、比较两个序列的方法
打点法只能大致了解两条序列的相似程度,而无法定量的描述。
4.1 打点法
4.1.1 基本概念和方法:

连续的对角线及对角线的平行线代表两条序列中相同的区域

可以通过自己对自己打点从而发现序列中重复的片段,这样搭打点的矩阵必然是对称的,并且有主对角线。
发现串联重复序列:
短串联重复序列:(STR) ,是一广泛存在于真核生物基因组中的DNA串联重复序列;遵循孟德尔显性遗传规律,广泛用于法医学个体识别、亲子鉴定等领域
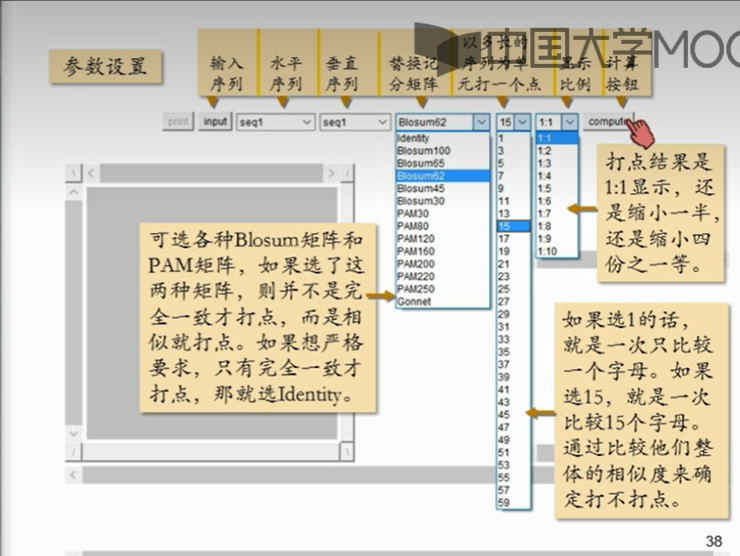
4.1.2 打点法在线软件:
Dotlet https://myhits.sib.swiss/cgi-bin/dotlet 基于Java开发
4.2序列对比法 alignment(对位排列,联配、对齐)
4.2.1 基本概念和方法
运用特定的算法找出两个和多个序列之间产生最大相似度得分的空格插入和序列对比方案。
举个例子:
通俗理解:通过插入空格,让两条序列相同的的字符尽可能的对应。

根据序列数量分为:多序列比对和双序列比对
双序列比对又分为全局比对和局部比对
4.2.2 双序列-全局比对算法
长度相似的两条序列适用于全局比对算法
动态规划算法 - Needleman-Wunsch算法
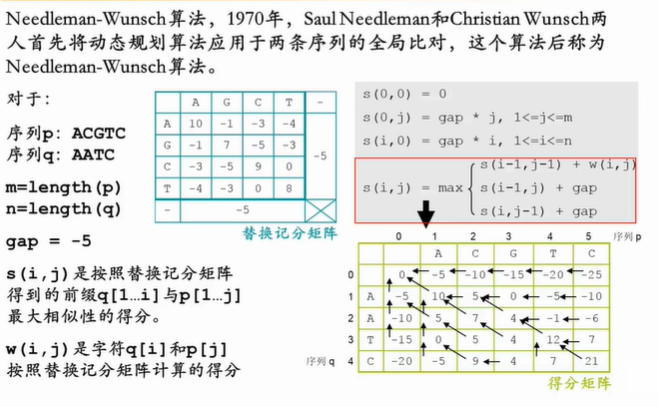
- 右下角的分数就是最终的得分
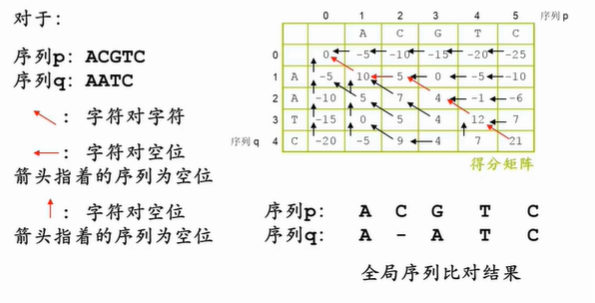
4.2.3双序列-局部对比算法
长度不同的序列(一长一短)两条序列适用于局部比对算法

相比与全局比对算法,多一个0的比较 为0时不用花箭头
局部比对的最终得分时得分矩阵中最大值(与全局比对不同)
从最大值处开始追溯
4.3 一致度与相似度的计算








